
二次有机气溶胶估算方法研究进展
2014-05-02 11:03:14闫才青李小滢王雪松张远航
中国环境科学 2014年3期
郑 玫,闫才青,李小滢,王雪松,张远航
(北京大学环境科学与工程学院,环境模拟与污染控制国家重点联合实验室,北京 100871)
二次有机气溶胶估算方法研究进展
郑 玫,闫才青,李小滢,王雪松,张远航*
(北京大学环境科学与工程学院,环境模拟与污染控制国家重点联合实验室,北京 100871)
我国区域复合污染日趋严重,二次有机气溶胶(SOA)是细颗粒物PM2.5的重要组成之一.本研究总结了当前国内外定量估算SOA的主要方法,包括EC示踪法、WSOC法、受体模型法、示踪物产率法、数值模拟法等.通过对各种方法的原理介绍与比较,指出1)EC示踪法、WSOC法、受体模型法简单易行,与在线连续观测数据结合可估算高时间分辨率的 SOA浓度,但受限于对当地源谱和特定物种的了解;2)示踪物产率法虽分析技术难度高,但可针对不同来源的SOA估算;3)数值模拟可获得大尺度SOA的空间分布.针对国内外的最新研究成果进行简单概述,不同方法的研究表明中国二次有机气溶胶是总有机气溶胶的重要部分,人为源VOCs对二次有机气溶胶发挥着重要贡献.本文旨在为未来我国二次有机气溶胶的研究提供基础信息和研究思路.
二次有机气溶胶;细颗粒物;定量估算方法;来源;中国
有机气溶胶(OA)包括污染源直接排放的一次有机气溶胶(POA),以及二次转化形成的二次有机气溶胶(SOA).SOA是由天然源VOCs和人为源 VOCs气态前体物与大气中的氧化剂(如O3、OH、NO3)发生光化学反应后,生成的半挥发和难挥发性有机物,经过气-粒转化或凝结到已有颗粒物上形成的有机气溶胶[1-2].
研究表明,SOA是大气气溶胶的重要组成部分.在美国东南部和加拿大,大约 10%~80%的有机气溶胶为二次来源[3-4];在中国,颗粒物中二次来源的有机物约占总有机物的 30%~95%[5-7].由于含氧、含氮等极性官能团的引入,二次有机气溶胶具有更强的极性和吸湿性,对能见度降低、灰霾形成、气候变化具有重要影响.
然而,由于对SOA复杂的大气形成过程、凝结/分配机制以及化学组成尚缺乏全面的认识,目前还不具备统一的对 SOA直接测量的分析手段,SOA的识别及其来源贡献的研究仍是国际气溶胶研究领域的热点和难点[8-9].除利用烟雾箱在特定的条件下直接模拟SOA的生成外,大气环境中SOA浓度的估算一般均采用间接方法.目前常用的SOA估算方法大致可概括为两大类,即基于观测数据的估算与基于源排放清单的模拟.前者主要包括EC示踪法、WSOC法、受体模型法、示踪物-产率法;而后者主要指数值模拟法.本文综述了国内外SOA定量估算的主要方法,总结国内SOA的最新研究结果,以期为评估我国SOA对细颗粒物的贡献以及相关控制对策的制定提供依据.
1 基于观测数据的估算法
1.1 EC示踪法
元素碳EC来自化石燃料和生物质等含碳燃料不完全燃烧的直接排放;一次燃烧排放 EC的同时也伴随产生有机碳OC.因此,EC可作为指示燃烧排放OC的示踪物种[3,10],根据EC可估算一次有机碳(POC),二次有机碳(SOC)则可经由式(1)、(2)计算得到[11]∶
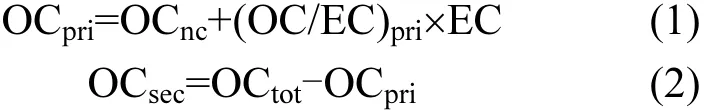
其中,OCpri, OCnc, (OC/EC)pri, OCsec, OCtot分别代表一次来源的有机碳、一次非燃烧源有机碳、一次燃烧源排放的OC与EC比值、二次来源的有机碳和测量的总有机碳.EC与OCtot可通过野外在线观测或实验室离线测量获取,常用仪器包括美国Sunset实验室的半连续或离线OC/EC分析仪(Sunset Laboratory Inc., Model 3型)、以及美国沙漠研究所研制的 OC/EC分析仪(DRI Model 2001型).
EC示踪法亦称为排放清单OC/EC比值法或最小OC/EC比值法[12],该法的关键在于OCnc与(OC/EC)pri的估算. (OC/EC)pri的获取主要通过以下几种方式∶(1)各种一次源排放清单中OC/EC比值的平均值[13];(2)将光化学反应较弱(光照强度弱、臭氧浓度低)、一次源排放占主导的相对稳定的大气环境下,环境样品的OC、EC数据做线性相关分析[11],回归曲线的截距即为OCnc,斜率则被视为(OC/EC)pri[14],OCnc通常被忽略不计[11];(3)观测期间环境OC与EC最小观测比值[15],即利用统计学方法,对观测所得的OC/EC比值做频率分布,选择一定置信区间(一般为5%或10%)的最小比值作为一次源排放的OC/EC比值[16].
EC示踪法由于仅需OC与EC数据,方法简单而被广泛应用[16-24].Cao等[18]在全国14个城市站点进行冬、夏季同步观测,利用EC示踪法估算表明中国城市大气环境中 SOC约占总碳的23.7%~41.5%; Zhang等[19]对中国18个城市、乡村、远郊站点长达一年的 OC、EC数据分析表明,城市与乡村大气中分别有 48%~62%和53%~83%的OC来自于二次转化.
基于EC示踪法开展的中国SOC研究(见图1)表明∶在空间分布上,北京[5]、天津[20]、济南[21]、厦门[22]等地 SOC浓度明显高于珠三角[23]、福建[24]地区,内陆城市高于沿海城市,北方高于南方;从季节变化上,各大城市秋、冬季SOC浓度明显高于春、夏季,且冬季最高,这可能是由于冬季供暖燃煤等人为源排放增加,半挥发性及挥发性SOA前体物排放增加[5],且扩散条件差,降水量少,湿去除不明显,较低的温度也有利于气-粒转换的发生[21].此外,污染物的区域传输[23]和气象条件(尤其是风向[22-23])均会影响二次有机污染物的分布.
EC示踪法适用于污染源种类和源排放量相对稳定的条件.然而,实际环境大气常处于动态变化中,即使取最小的OC/EC比值仍难以避免二次有机物的干扰[12];同时,气象条件、长距离传输等都会影响OC/EC比值[22]. 借助高时间分辨率的CO数据对OC进行标准化,可减少因新鲜排放、稀释、大气混合等导致的OC变化[25-26]. Hu等[27]利用统计方法,先假定OC/EC比值范围,以0.001为步长选择不同 OC/EC比值作为(OC/EC)pri,分别用EC示踪法求解,获得的各组SOC与实测EC相关系数最小时对应的 OC/EC比值被视为(OC/EC)pri;Yu等[28]利用排放/传输模型(如多尺度综合空气质量模型,CMAQ),结合高时间分辨率排放清单,追踪模拟(OC/EC)pri,并与测量获取的环境OC、EC数据结合,为区分POC和SOC提供了新思路.此外,不同OC、EC分析方法(热光反射法 TOR和热光透射法 TOT)会导致测量的OC/EC比值不同[29],增加估算结果的不确定性.利用环境OC与EC浓度估算SOC时需评估分析方法的差异,尽量获取接近一次燃烧源的OC/EC比值.
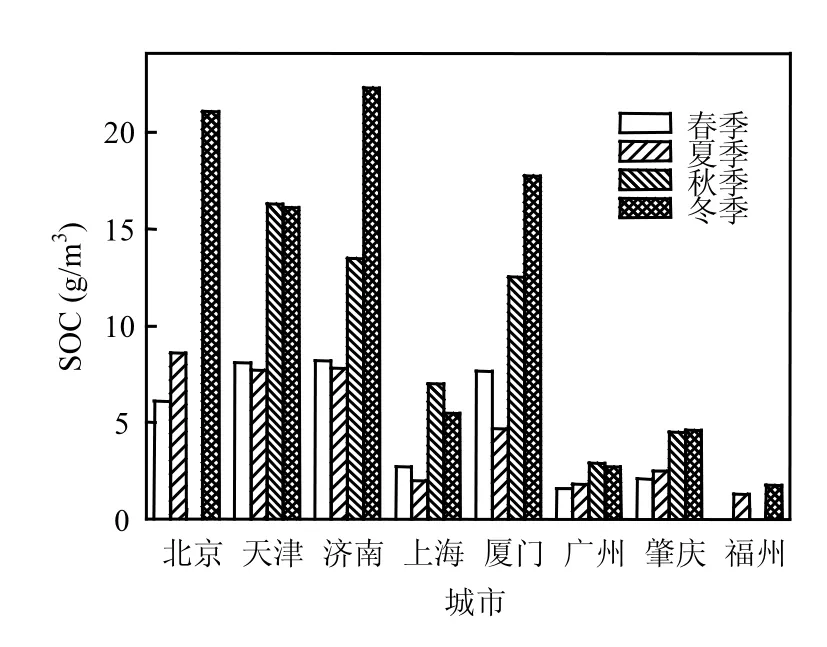
图1 中国各大城市基于EC示踪法的SOC浓度分布[5,20-24]Fig.1 SOC mass concentration in different cities of China based on the EC tracer method[5,20-24]
在 EC示踪法的基础上,一些研究者利用其他物种(如CO)代替EC作为一次燃烧源示踪物,或同时采用多个物种加以限制(如多元回归法)[25-26,30],引申了 EC示踪法.如 Blanchard等[26]利用CO和EC作为POC指示物,SO42-、NO3-和臭氧作为 SOC指示物,通过多元回归法,估算POC与SOC. Pachon等[30]在Blanchard基础上增加K+指示生物质燃烧排放的POC,与CO、EC共同作为总POC的指示物.
1.2 WSOC法
水溶性有机物(WSOC)是总有机碳的重要组分部分.大气颗粒物中WSOC主要来自生物质燃烧排放[31]和二次转化[32].总 WSOC扣除生物质燃烧贡献即可估算二次转化来源的WSOC;而在生物质燃烧可忽略的地区,WSOC被用来指示二次有机碳[33].
生物质燃烧产生的WSOC可通过测量该排放源的示踪物左旋葡聚糖(levoglucosan)来推算
[34].较 K+而言,左旋葡聚糖对生物质燃烧的指示更为独特,且在有机溶剂可提取的有机物种中丰度高,因而是生物质燃烧源较为理想的示踪物选择.此方法的原理如式(3)、(4)所示∶
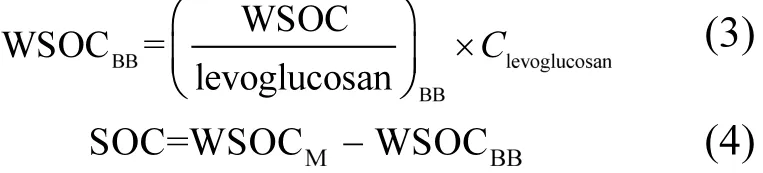
式中∶WSOCM表示测量的总 WSOC浓度; WSOCBB与(WSOC/levoglucosan)BB分别代表生物质燃烧源排放的WSOC浓度及WSOC与左旋葡聚糖的浓度比值,而 Clevoglucosan则为环境中左旋葡聚糖的浓度.
WSOC法所需检测的物种相对较少,在实际应用中简单易行.利用在线水萃取装置(Particle into Liquid Sampler, PILS)[33]或在线气体-气溶胶收集仪(Online Gas and Aerosol Collector, GAC)连接TOC分析仪,可获取连续在线的WSOC浓度,从而提供高时间分辨率SOC估算的基础数据.
WSOC法假设WSOC仅来自于二次转化和生物质燃烧,忽略了 WSOC的其他一次来源,如机动车排放[35].方法中(WSOC/levoglucosan)BB需借助当地生物质燃烧源的源谱获取,该比值具有明显的地区差异性,且随燃烧生物质的种类和燃烧条件等不同.例如,美国亚特兰大的研究[36-37]显示,计划性生物质燃烧产生的(WSOC/ levoglucosan)BB约为9.1±2.9,而野火燃烧排放产生的比值约为 4.5~6.8.此外,WSOC法忽略了部分水溶性较低的 SOC贡献,可能会导致对 SOC的低估.
1.3 受体模型法
化学质量平衡模型(Chemical Mass Balance, CMB)、正定矩阵分解模型(Positive Matrix Factorization, PMF)是目前大气颗粒物来源研究中广泛应用的两种受体模型.二者主要用于解析一次源对受体点颗粒物的浓度贡献,但近几年也逐渐与其它手段结合同时解析一次与二次源对颗粒物或有机碳的浓度贡献.
1.3.1 CMB-MM 法 化学质量平衡模型结合分子示踪物(CMB-MM)法是在CMB中利用有机分子作为源的示踪物种定量各种源对颗粒物或OC贡献的方法[4].在解析OC来源的研究中,解析结果中由一次源贡献的OC之和视为POC,总OC与POC之差可间接作为SOC[38],估算如下∶
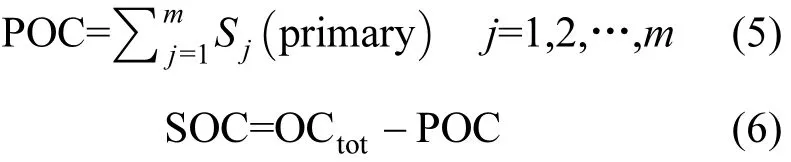
其中,Sj代表一次源j对OC浓度的贡献,OCtot代表测量的总OC.
CMB-MM模型法解析OC来源时输入数据包括受体点有机示踪物种的浓度及其不确定性,以及污染源成分谱;通常采用有效方差最小二乘法确定各污染源的贡献值.污染源成分谱则尽可能应用当地的源成分谱,美国已建立了可共享的挥发性有机物和颗粒物的污染源成分谱数据库 SPECIATE,然而,我国源谱工作的开展地区及研究的源谱种类均相对稀少,且主要集中在北方及东部沿海发达地区,如北京、天津、济南、南京等城市已针对燃煤源、机动车源、尘源(土壤尘、道路尘)等源类建立了当地的源成分谱,而冶炼源、生物质燃烧、餐饮源等源类的研究较为有限[39].
除一次污染源成分谱外,部分研究尝试在CMB模型里引入虚拟的“纯”OC源成分谱,作为SOC源谱,解析获得SOC和其他一次源类的贡献[40].Shi等[41]基于 CMB模型结果,通过嵌套的迭代估算方法扣除SOC,并将扣除全部SOC后的受体成分谱与源成分谱纳入 CMB模型,估算POC与SOC浓度,及各个源的贡献值.CMB-MM法建立在所有一次污染源类均被鉴别的基础上,若CMB模型的输入源谱缺少某些POC贡献源类,则会导致对SOC的高估.
14C同位素示踪技术可以区分现代生物源和化石燃料源对大气含碳颗粒物的贡献[42].主要原理基于大气14CO2中的14C通过光合作用可储存在新形成的含碳物质中;而化石燃料的形成时间远远大于14C的半衰期(约5730年),化石燃料中不存在14C.通过测量14C,可定量估算颗粒物中来自化石燃料碳和现代碳的贡献.Ding等[42]利用14C技术解析颗粒物中总碳的现代源与化石燃料源的贡献量,并将CMB-MM模型解析结果中木柴燃烧、餐饮、植物碎屑等源类统归为一次现代源,柴油车、汽油车、焦化厂等源类划分为一次化石燃料源;总碳与一次源碳解析结果中两类源的贡献量分别做差值,即对应SOC中现代源和化石燃料源的贡献.
1.3.2 AMS-PMF法 与CMB法不同,PMF法是基于统计分析的方法,需输入大量颗粒物化学物种浓度数据,以及各化学物种的测量偏差;但不需源成分谱的详细信息,避免了源谱测试本身的不确定性,以及借用外地源谱而导致的估算结果偏差.利用PMF模型可解析出颗粒物的一次源类因子贡献,若将分配到具有二次来源特征因子(如硫酸盐、硝酸盐等)中的有机碳判定为 SOC,则可简单估算SOC的浓度.Yuan等[43]利用该法估算香港SOC平均浓度为4.25µgC/m3,约占香港年均总OC的46%.
近年来气溶胶质谱技术在大气环境领域得到广泛应用.AMS(Aerosol Mass Spectrometer, Aerodyne Inc., USA)可提供高时间分辨率的超细颗粒物PM1的化学成分信息,满足PMF需大量数据的要求.通过筛选 AMS提供的高质量分辨率有机气溶胶离子碎片(m/z约1~300),输入PMF受体模型进行来源解析;由 AMS有机气溶胶质谱图的时间序列,即单位时间分辨率内大气颗粒物平均有机气溶胶质谱图,可分解出多个有机气溶胶因子/源类质谱图、各因子/源类贡献浓度,可区分颗粒物中代表一次排放的类似烃类有机气溶胶(Hydrocarbon-like Organic Aerosol, HOA)和二次生成的含氧有机气溶胶(Oxygenated Organic Aerosol, OOA)的贡献[44-45].
AMS质谱图中较为重要的有机碎片包括m/z 43、m/z 44、m/z 55和m/z 57,通过外场观测和烟雾箱实验已证明这些碎片及其相对丰度可用于区分颗粒物源类.其中m/z 57 (C4H9+)通常来源于饱和直链烃类分子的断裂,可代表一次源排放的颗粒有机物;m/z 44 (CO2+)一般形成于羧基在电子轰击电离下的断裂,可认为来自于含氧有机物,而m/z 43 (C3H7+和C2H3O+)可来自于一次来源和二次/氧化来源的贡献[46].
黄晓峰等[9]利用 AMS-PMF法解析出深圳冬季大气细粒子的 5类因子,将其中对一次有机气溶胶特征离子碎片峰贡献大的因子中有机物加和作为POA,而将分配到具有二次来源特征因子(如SO42-、NO3-等)中的有机物加和作为SOA,发现深圳冬季 SOA 浓度水平约为(9.41± 6.33)µg/m3,占总有机物质量的40%,与POA相比空间上浓度变化小,体现了区域性二次污染物特征.Xiao等[47]利用AMS-PMF法估算表明PRD地区夏季SOA约占总有机物的53%~66%.
此外,傅立叶变换红外 FTIR技术可通过对有机官能团进行定性与定量来估算 SOA[48]. FTIR提供的官能团信息与AMS有机离子碎片的PMF因子解析,对比表明两种方法在SOA源类型的识别、各源/因子的质量分数和时间序列等方面都具有良好的一致性[49].
1.4 示踪物-产率法
除上述针对SOA总量的估算方法外,近年来SOA来源研究成为控制二次有机污染的关键.异戊二烯和单萜烯是自然界中排放量较高的两种天然源VOCs,挥发性芳香族化合物则是重要的人为源VOCs.近年来,烟雾箱模拟实验中异戊二烯、单萜烯、倍半萜烯和芳香烃等VOCs的氧化产物在外场观测中被检测到[50-51].美国环保署(USEPA)建立了一种利用大气颗粒物中可检测的VOCs氧化产物作为二次有机示踪物来估算人为源及天然源SOA的方法,即示踪物-产率法[52].
该方法基于烟雾箱实验,首先通过GC/MS测定每种VOC前体物在烟雾箱中反应生成的一系列有机示踪物的浓度,将所有SOA有机示踪物的浓度之和,除以烟雾箱反应后产生的总气溶胶(即总 SOA)的浓度,即为SOA的质量分数(FSOA),见式(7).

利用烟雾箱获得的OM/OC比值,经过SOA与SOC的转换,转换为SOC质量分数(FSOC)[52],并假定烟雾箱实验中生成的二次有机示踪物与对应前体物产生SOA的质量关系与实际大气中形成的相同[52-53].从而,通过测定实际大气中各种前体物产生的SOA示踪物浓度借助FSOA(FSOC)可估算某一特定 VOC前体物产生的总 SOA (SOC)浓度,并推算不同来源 SOA(SOC)对 OA (OC)的贡献.
目前烟雾箱实验获得的异戊二烯、α-蒎烯、β-丁香烯、甲苯等前体物对应的 FSOC值[54]已被应用于广州[6]、北京[7]、香港[54]3个超大城市SOC的估算.研究表明,人为源VOCs前体物对北京和广州等地(不论城、郊)SOC均有重要贡献;广州人为源芳香烃前体物对 SOC的贡献高达79%,较北京(45%~54%)更为显著,均远高于以天然源VOCs贡献为主的香港地区.
国内对SOA示踪物-产率法的应用较少,除了上述研究外,Fu等[55]测定了泰山上以α/β-蒎烯和 β-丁香烯为前体物的 SOA浓度,分别可占到总有机物的 10%和 9.8%.目前,该法主要考虑了以异戊二烯、单萜烯、倍半萜烯等天然源VOCs为前体物,以及甲苯等人为源芳香烃为前体物产生的 SOA.然而,仅仅考虑这几种主要的前体物可能导致 SOA含量的低估.此外,该法忽略了云中过程形成的氧化性产物(如乙二醛等),这些产物分配到云滴、雾、湿气溶胶中,经过水相反应,转化为羧酸类物质,具有较强的 SOA生成潜力
[56].未来的研究应侧重于增加和完善对人为源前体物的二次有机示踪物的定性与定量研究,确定更符合实际大气环境的SOA质量分数,减少示踪物-产率法的不确定性.
2 基于排放清单的数值模拟法
空气质量模型(CMAQ、GEOS-CHEM、CAMx等)也逐渐被用于SOA的模拟,成为研究大尺度 SOA空间分布和源贡献的一个重要手段和热点方向[57].与受体模型估算 SOA的方法不同,空气质量模型利用烟雾箱实验获得的SOA产率、气-粒分配系数等信息建立 SOA模块[25],在输入准确的源排放清单和气象资料条件下,通过对影响SOA浓度水平的各种大气过程(如VOCs前体物的排放和氧化过程、半挥发性有机物的气-粒分配过程,以及传输、扩散、沉降等)的模拟,提供SOA浓度的时空变化及其与排放源之间的关系.
空气质量模式对SOA浓度的模拟值往往低于观测值,其中一个重要原因是模式中对SOA反应机制及前体物的了解尚有待完善.早期的CMAQ版本仅将单萜烯、长链烷烃和芳香烃视为SOA来源[58],且未考虑在酸性和高NOx条件下 SOA的产率和生成速率的变化等[1];CMAQ 4.7版本考虑了芳香烃类VOCs前体物在氮氧化物存在下的SOA产率,增加了苯、异戊二烯、倍半萜烯等SOA前体物,以及乙二醛和丙酮醛的云中氧化反应、颗粒态的低聚反应、酸对异戊二烯生成SOA的催化作用等机制[59].GEOS-Chem模型可用于模拟由生物源萜烯、异戊二烯,以及人为活动和生物质燃烧排放的芳香烃类等产生的半挥发性含氧有机物通过平衡分配产生的SOA[60],同时,还用于模拟乙二醛、丙酮醛的水相反应生成的SOA[61];近年来,GEOS-Chem模型中已增加了二羰基化合物、半挥发性和中间体VOCs生成SOA的机制[62-63].
此外,减少SOA前体物源排放清单的不确定性[63],填补模式中有机气溶胶缺失的重要源、细化源清单的空间、时间分辨率,都会使得模拟结果更能够接近实测值.
程艳丽等[64]利用二维空气质量模式,对珠三角区域尺度SOA进行了模拟研究,表明SOA的生成具有明显的光化学反应特征,浓度高值出现在14∶00左右.Fu等[62]利用GEOS-Chem模型模拟中国大陆SOA浓度,表明中国地面21%的OC来自于二次源,而东部地区夏季高达 62%的 OC为二次来源;并指出中国最重要的SOC前体物是异戊二烯、单萜烯和芳香烃.Jiang等[65]利用WRF/Chem结合SOA模型(SORGAM)研究表明中国人为源对总SOA的贡献约为35%,春、夏、秋、冬各季分别为41%、26%、39%、59%;模拟值对SOA浓度低估约为0~75%.
3 各种估算方法的比较
SOA的估算方法多样,已从SOA总量的估算发展到针对各种特定前体物产生SOA浓度的细化估算.但现有方法各有利弊,仍缺乏普遍适用的“统一”方法(表1).
总的来说,基于观测数据的估算方法,如 EC示踪法、WSOC法、AMS-PMF法等,简单易行,可结合高时间分辨率的在线数据,快速估算环境大气中 SOC(SOA)的总体浓度;但由于分析物种有限,未能建立 SOC(SOA)浓度与源之间的关系.相比之下,14C技术结合受体模型CMB-MM法可区分化石燃料源和现代生物源 SOA,示踪物-产率法则可针对性地评估某一类特定 VOCs前体物对 SOA的浓度贡献,而受限于烟雾箱实验对VOCs光化学产物的准确认识,及SOA示踪物的测量分析困难,示踪物-产率法仍未得到广泛的应用.
基于观测数据的估算方法依赖于外场观测的时间跨度和空间尺度,往往只能反映某一或某些观测点位的大气环境;而基于源清单的数值模拟估算可覆盖较大的空间范围,据某地区或某区域的源排放清单和气象场,可进行大尺度范围、高时间分辨率SOA浓度模拟及来源解析;空气质量模型模拟的准确与否,与源清单、气象场和SOA生成模块完善程度紧密相关.
目前的研究多采用单一的估算方法,针对多种方法的对比研究相对较少.Pachon等[30]对 EC示踪法、回归法、CMB模型法、PMF模型法等对SOA的估算结果进行了较为综合的比较,发现CMB模型评估结果最高(SOC占 OC的 46%), PMF模型评估结果最低(占OC的26%);而根据误差传递、季节评估、日变化研究等对各种估算方法的不确定性和估算结果进行比较发现,回归法具有最小的不确定性;在进行长时间序列SOC的估算时,回归法具有优势.
国内针对二次有机气溶胶的浓度水平、来源贡献的研究尚处于起步阶段.目前应用最为广泛的SOA估算方法是EC示踪法,其他方法的使用相对较少.Xiao等[47]初步尝试利用同一外场观测数据比较了EC示踪法、WSOC法和AMS-PMF法的估算结果,发现三种方法具有较高的一致性.Yuan等[43]在香港的研究中,对比表明EC示踪法所得夏、冬两季SOC较PMF模型法分别高估约 70%~212%和 4%~43%,主要是由于缺乏随时空变化的一次混合源OC/EC比值.Ding等[6]在珠三角的研究发现,夏季示踪物产率法与 EC-示踪法的估算结果虽不一致但相关性很好,而在生物质燃烧明显的秋冬季节,由于生物质燃烧源中OC/EC比值高,EC示踪法高估,明显高于示踪物-产率法.然而,目前国内外针对同一观测期间的多种方法(尤其是基于观测和污染源排放清单方法)的比较,以及对 OC质量浓度闭合的研究均十分缺乏.

表1 各种SOC估算方法比较Table 1 Comparison of different methods for estimating SOC levels
4 结语
在选择 SOA估算方法时,应根据研究地区所具备的各估算方法输入条件及所需达到的预期目标来综合考虑.对源清单较为完善的地区,可结合空气质量模型或源清单 OC/EC比值法进行估算;在源成分谱较完善的地区,可利用CMB-MM受体模型法或WSOC法开展;而在有机物种数据相对缺乏的地区,则可选择EC示踪法进行;在排放源以有机物贡献为主的地区,示踪物-产率法、AMS-PMF法可提供源类别多样化和高时间分辨率的 SOA信息.如果关注某区域大尺度内 SOA的浓度水平,则可采用数值模拟的手段来研究.未来的研究有待将多种 SOA的估算方法对比和综合利用,并对各种方法的不确定性进行评估.
[1] Jang M, Czoschke N M, Lee S, et al. Heterogeneous atmospheric aerosol production by acid-catalyzed particle-phase reactions [J]. Science, 2002,298:814-817.
[2] 谢绍东,于 淼,姜 明.有机气溶胶的来源与形成研究现状 [J].环境科学学报, 2006,26(12):1933—1939.
[3] Turpin B J, Huntzicker J J. Identification of secondary organic aerosol episodes and quantitation of primary and secondary organic aerosol concentrations during SCAQS [J]. Atmospheric Environment, 1995,29(23):3527-3544.
[4] Zheng M, Cass G R, Ke L, et al. Source apportionment of daily fine particulate matter at Jefferson Street, Atlanta, GA, during summer and winter [J]. Journal of the Air and Waste Management Association, 2007,57(2):228-242.
[5] Dan M, Zhuang G S, Li X X, et al. The characteristics of carbonaceous species and their sources in PM2.5in Beijing [J]. Atmospheric Environment, 2004,38(21):3443-3452.
[6] Ding X, Wang X M, Gao B, et al. Tracer-based estimation of secondary organic carbon in the Pearl River Delta, south China [J]. Journal of Geophysical Research-Atmospheres, 2012,117, D05313,doi:10.1029/2011JD016596.
[7] Guo S, Hu M, Guo Q F, et al. Primary sources and secondary formation of organic aerosols in Beijing, China [J]. Environmental Science and Technology, 2012,46(18):9846-9853.
[8] Rodrigo S A, Raul G E M S, Manuel A L G. Estimations of primary and secondary organic carbon formation in PM2.5aerosols of Santiago City, Chile [J]. Atmospheric Environment, 2009,43(13):2125-2131.
[9] 黄晓峰,赵倩彪,何凌燕,等.基于气溶胶质谱的二次有机气溶胶识别 [J]. 中国科学:化学, 2010,40(10):1550-1557.
[10] Cabada J C, Pandis S N, Subramanian R, et al. Estimating the secondary organic aerosol contribution to PM2.5using the EC tracer method [J]. Aerosol Science and Technology, 2004,38(51):140-155.
[11] Lim H J, Turpin B J. Origins of primary and secondary organic aerosol in Atlanta: results of time-resolved measurements during the Atlanta supersite experiment [J]. Environmental Science and Technology, 2002,36(21):4489-4496.
[12] 叶文媛,吴 琳,冯银厂,等.大气中二次有机气溶胶估算方法研究进展 [J]. 安全与环境学报, 2011,11(1):127-130.
[13] Gray H A, Cass G R, Huntzicker J J, et al. Characteristics of atmospheric organic and elemental carbon particle concentrations in Los Angeles [J]. Environmental Science and Technology, 1986,20(6):580-589.
[14] Saylor R D, Edgerton E S, Hartsell B E. Linear regression techniques for use in the EC tracer method of secondary organic aerosol estimation [J]. Atmospheric Environment, 2006,40(39):7546-7556.
[15] Na K, Sawant A A, Song C, et al. Primary and secondary carbonaceous species in the atmosphere of Western Riverside County, California [J]. Atmospheric Environment, 2004,38(9):1345-1355.
[16] Lin P, Hu M, Deng Z, et al. Seasonal and diurnal variations of organic carbon in PM2.5in Beijing and the estimation of secondary organic carbon [J]. Journal of Geophysical Research-Atmospheres, 2009,114,D00G11,doi:10.1029/2008JD010902.
[17] Turpin B J, Huntzicker J J, Larson S M, et al. Los Angeles summer midday particulate carbon: primary and secondary aerosol [J]. Environmental Science & Technology, 1991,25(10):1788-1793.
[18] Cao J J, Lee S C, Chow J C, et al. Spatial and seasonal distributions of carbonaceous aerosols over China [J]. Journal of Geophysical Research-Atmospheres, 2007,112,D22S11,doi:10. 1029/2006JD008205.
[19] Zhang X Y, Wang Y Q, Zhang X C, et al. Carbonaceous aerosol composition over various regions of China during 2006 [J]. Journal of Geophysical Research-Atmospheres, 2008,113, D14111,doi:10.1029/2007JD009525.
[20] Gu J X, Bai Z P, Liu A X, et al. Characterization of atmospheric organic carbon and element carbon of PM2.5and PM10at Tianjin, China [J]. Aerosol and Air Quality Research, 2010,10:167-176.
[21] Yang L X, Zhou X H, Wang Z, et al. Airborne fine particulate pollution in Jinan, China: Concentrations, chemical compositions and influence on visibility impairment [J]. Atmospheric Environment, 2012,55:506-514.
[22] Zhang F W, Zhao J P, Chen J S, et al. Pollution characteristics of organic and elemental carbon in PM2.5in Xiamen, China [J]. Journal of Environmental Sciences, 2011,23(8):1342-1349.
[23] Huang H, Ho K F, Lee S C, et al. Characteristics of carbonaceous aerosol in PM2.5: Pearl Delta River Region, China [J]. Atmospheric Research, 2012,104-105:227-236.
[24] Zhang F W, Xu L L, Chen J S, et al. Chemical characteristics of PM2.5during haze episodes in the urban of Fuzhou, China [J]. Particuology, 2012,11(3):264-272.
[25] Lee S, Wang Y H, Russell A G. Assessment of secondary organic carbon in the southeastern United States: A review [J]. Journal of the Air and Waste Management Association, 2010,60(11):1282-1292.
[26] Blanchard C L, Hidy G M, Tanenbaum S, et al. Carbon in southeastern U.S. aerosol particles: Empirical estimates of secondary organic aerosol formation [J]. Atmospheric Environment, 2008,42(27):6710-6720.
[27] Hu W W, Hu M, Deng Z Q, et al. The characteristics and origins of carbonaceous aerosol at a rural site of PRD in summer of 2006 [J]. Atmospheric Chemistry and Physics, 2012,12:1811-1822.
[28] Yu S C, Dennis R L, Bhave P V, et al. Primary and secondary organic aerosols over the United States: estimates on the basis of observed organic carbon (OC) and elemental carbon (EC), and air quality modeled primary OC/EC ratios [J]. AtmosphereEnvironment, 2004,38(31):5257-5268.
[29] Cheng Y, Zheng M, He K B, et al. Comparison of two thermal-optical methods for the determination of organic carbon and elemental carbon: Results from the southeastern United States [J]. Atmosphere Environment, 2011,45(11):1913-1918.
[30] Pachon J E, Balachandran S, Hu Y T, et al. Comparison of SOC estimates and uncertainties from aerosol chemical composition and gas phase data in Atlanta [J]. Atmospheric Environment, 2010,44(32):3907-3914.
[31] Mukai H, Ambe Y. Characterization of a humic acid-like brown substance in airborne particulate matter and tentative identification of its origin [J]. Atmospheric Environment, 1986, 20(5):813-819.
[32] Saxena P, Hildemann L M. Water-Soluble organics in atmospheric particles: A critical review of the literature and application of thermodynamics to identify candidate compounds [J]. Journal of Atmospheric Chemistry, 1996,24(1):57-109.
[33] Weber R J, Sullivan A P, Peltier R E, et al. A study of secondary organic aerosol formation in the anthropogenic-influenced southeastern United States [J]. Journal of Geophysical Research-Atmosphere, 2007,112,D13302,doi:10.1029/2007JD008408.
[34] Fraser M P, and Lakshmanan K. Using levoglucosan as a molecular marker for the long-range transport of biomass combustion aerosols [J]. Environmental Science and Technology, 2000,34(21):4560-4564.
[35] Fisseha R, Dommen J D, Gaeggeler K, et al. Online gas and aerosol measurement of water soluble carboxylic acids in Zurich [J]. Journal of Geophysical Research-Atmospheres, 2006,111, D12316,doi:10.1029/2005JD006782.
[36] Yan B, Zheng M, Hu Y T, et al. Organic composition of carbonaceous aerosols in an aged prescribed fire plume [J]. Atmospheric Chemistry and Physics, 2008,8:6381-6394.
[37] Yan B. Characterization and source apportionment of ambient PM2.5in Atlanta, Georgia: On-road emission, biomass burning and SOA impact [D]. United States: Georgia Institute of Technology, 2009.
[38] Zheng M, Ke L, Edgerton E S, et al. Spatial distribution of carbonaceous aerosol in the southeastern United States using molecular markers and carbon isotope data [J]. Journal of Geophysical Research-Atmospheres, 2006,111,D10S06,doi:10.1029/2005JD006777.
[39] 郑 玫,张延君,闫才青,等.上海 PM2.5工业源谱的建立 [J]. 中国环境科学, 2013,33(8):1354-1359.
[40] Lee S, Liu W, Wang Y H, et al. Source apportionment of PM2.5:comparing PMF and CMB results for four ambient monitoring sites in the southeastern United States [J]. Atmospheric Environment, 2008,42(18):4126-4137.
[41] Shi G L, Tian Y Z, Zhang Y F, et al. Estimation of the concentrations of primary and secondary organic carbon in ambient particulate matter: application of the CMB-iteration method. Atmospheric Environment, 2011,45:5692-5698.
[42] Ding X, Zheng M, Edgerton E S, et al. Contemporary or fossil origin: Split of estimated secondary organic carbon in the southeastern United States [J]. Environmental Science and Technology, 2008,42(24):9122-9218.
[43] Yuan Z B, Yu J Z, Lau A K H, et al. Application of positive matrix factorization in estimating aerosol secondary organic carbon in Hong Kong and its relationship with secondary sulfate [J]. Atmospheric Chemistry and Physics, 2006,6:25-34.
[44] Huang X F, He L Y, Hu M, et al. Highly time-resolved chemical characterization of atmospheric submicron particles during 2008Beijing Olympic Games using an Aerodyne High-Resolution Aerosol Mass Spectrometer [J]. Atmospheric Chemistry and Physics, 2010,10(18):8933-8945.
[45] Sun Y L, Zhang Q, Anastasio C, et al. Insights into secondary organic aerosol formed via aqueous-phase reactions of phenolic compounds based on high resolution mass spectrometry [J]. Atmospheric Chemistry and Physics, 2010,10(10):4809-4822.
[46] Zhang Q, Worsnop D R, Canagaratna M R, et al. Hydrocarbonlike and oxygenated organic aerosols in Pittsburgh: insights into sources and processes of organic aerosols [J]. Atmosphere Chemistry and Physics, 2005,5(12):3289-3311,doi:10.5194/acp-5-3289-2005.
[47] Xiao R, Takegawa N, Zheng M, et al. Characterization and source apportionment of submicron aerosol with aerosol mass spectrometer during the PRIDE-PRD 2006campaign [J]. Atmospheric Chemistry and Physics, 2011,11(14):6911-6929.
[48] Maria S F, Russell L M, Turpin B J, et al. FTIR measurements of functional groups and organic mass in aerosol samples over the Caribbean [J]. Atmospheric Environment, 2002,36(33):5185-5196.
[49] Liu S, Ahlm L, Day D A, et al. Secondary organic aerosol formation from fossil fuel sources contribute majority of summertime organic mass at Bakersfield [J]. Journal of Geophysical Research, 2012, 117,D00V26,doi:10.1029/2012JD018170.
[50] Jaoui M, Kleindienst T E, Lewandowski M, et al. Identification and quantification of aerosol polar oxygenated compounds bearing carboxylic or hydroxyl groups. 2. Organic tracer compounds from monoterpenes[J]. Environmental Science and Technology, 2005,39(15):5661-5673.
[51] Szmigielski R, Surratt J D, Gómez-González Y, et al. 3-methyl-1,2,3-butanetricarbocylic acid: An atmospheric tracer for terpene secondary organic aerosol [J]. Geophysical Research Letters, 2007,34(24),L24811,doi:10.1029/2007GL031338.
[52] Kleindienst T E, Jaoui M, Lewandowski M, et al. Estimates of the contributions of biogenic and anthropogenic hydrocarbons to secondary organic aerosol at a southeastern US location [J]. Atmospheric Environment, 2007,41(37):8288-8300.
[53] Kleindienst T E, Jaoui M, Lewandowski M, et al. The formation of SOA and chemical tracer compounds from the photoxidation of naphthalene and its methyl analogs in the presence and absence of nitrogen oxides [J]. Atmospheric Chemistry and Physics, 2012,12,8711-8726.
[54] Hu D, Bian Q J, Li T W Y, et al. Contributions of isoprene, monoterpenes, β-caryophyllene, and toluene to secondary organic aerosols in Hong Kong during the summer of 2006 [J]. Journal of Geophysical Research, 2008,113,D22206,doi:10.1029/2008JD010437.
[55] Fu P Q, Kawamura K, Kanaya Y, et al. Contributions of biogenic volatile organic compounds to the formation of secondary organic aerosols over Mt. Tai, Central East China [J]. Atmospheric Environment, 2010,44(38):4817-4826.
[56] Lim Y B, Tan Y, Turpin B J. Chemical insights, explicit chemistry and yields of secondary organic aerosol from methylglyoxal and glyoxal [J]. Atmospheric Chemistry and Physics Discussion, 2013,13,4687-4725.
[57] Lane T E, Donahue N M, Pandis S N. Simulating secondary organic aerosol formation using the volatility basis-set approach in a chemical transport model [J]. Atmospheric Environment, 2008,42(32):7439-7451.
[58] Binkowski F S, Roselle S J. Model-3community multiscale air quality (CMAQ) model aerosol component 1. Model description [J]. Journal of Geophysical Research-Atmospheres, 2003,108,D6,4183,doi:10.1029/2001JD001409.
[59] Carlton A G, Bhave P V, Napelenok S L, et al. Model representation of secondary organic aerosol in CAMQv4.7 [J]. Environmental Science & Technology, 2010,44(22):8553-8560.
[60] Henze D K, Seinfeld J H, Ng N L, et al. Global modeling of secondary organic aerosol formation from aromatic hydrocarbons:high-vs. low-yield pathways [J]. Atmospheric Chemistry and Physics, 2008,8:2405—2421.
[61] Fu T M, Jacob D J, Heald C L. Aqueous-phase reactive uptake of dicarbonyls as a source of organic aerosol over eastern North America [J]. Atmospheric Environment, 2009,43(10):1814—1822.
[62] Fu T M, Cao J J, Zhang X Y, et al. Carbonaceous aerosols in China: top-down constraints on primary sources and estimation of secondary contribution [J]. Atmospheric Chemistry and Physics, 2012,12(5):2725-2746.
[63] Pye H O T and Seinfeld J H. A global perspective on aerosol from low-volatility organic compounds [J]. Atmospheric Chemistry and Physics, 2010,10:4377—4401.
[64] 程艳丽,李湉湉,白郁华,等.珠江三角洲区域大气二次有机气溶胶的数值模拟 [J]. 环境科学, 2009,30(12):3441-3447.
[65] Jiang F, Liu Q, Huang X X, et al. Regional modeling of secondary organic aerosol over China using WRF/Chem [J]. Journal of Aerosol Science, 2012,43(1):57-73.
[66] Pun B K, Wu S Y, Seigneur C, et al. Uncertainties in modeling secondary organic aerosols: Three-dimensional modeling studies in Nashville/Western Tennessee [J]. Environmental Science and Technology, 2003,37(16):3647-3661.
《中国环境科学》被Ei收录
根据 Ei总部 2013年颁布的期刊收录情况,《中国环境科学》已被 Ei数据库作为源期刊收录,详见http://www.chinaeidata.com/periodical.htm
《中国环境科学》编辑部
2013-03-14
A review of methods for quantifying secondary organic aerosol.
ZHENG Mei, YAN Cai-qing, LI Xiao-ying, WANG Xue-song, ZHANG Yuan-hang*
(State Key Joint Laboratory of Environmental Simulation and Pollution Control, College of Environmental Sciences and Engineering, Peking University, Beijing 100871, China). China Environmental Science, 2014,34(3):555~564
Regional air pollution is complex and becomes increasingly important in China. Among many others, secondary organic aerosol (SOA) is one of the most important components of PM2.5. This paper discusses various methods for quantifying SOA in the atmosphere (including methods based on the EC tracer, WSOC, receptor model, the SOA tracers, and air quality model), presents the basic principle of each method and points out that 1) the EC-tracer method, the WSOC method and the receptor model method are relatively simple and convenient, but limited by the availability of local source profiles and some specific tracers; 2) the SOA-tracer method is analytically challenging but can supply source-specific SOA information; and 3) the air quality model method can provide large scale spatial distribution of SOA. This paper also summarizes the most recent results of SOA research in China and abroad and indicates that SOA is important in organic aerosol, and anthropogenic VOCs play a significant role in SOA formation in China. The primary purpose of this review is to provide basic and integrated information and suggestion for future directions of SOA study in China.
secondary organic aerosol;fine particulate matter;quantification method;source;China
X513
:A
:1000-6923(2014)03-0555-10
郑 玫(1969-),女,贵州贵阳人,教授,从事一次和二次来源的有机气溶胶物化特性及大气颗粒物来源解析的研究工作.发表论文65篇.
2013-05-03
国家自然科学基金资助项目(21190050,41175102);上海市环保科研项目(45210128-0432)
* 责任作者, 教授, yhzhang@pku.edu.cn
猜你喜欢
湘潮(上半月)(2021年10期)2021-12-02 02:09:38
能源工程(2021年5期)2021-11-20 05:50:44
生物质化学工程(2021年1期)2021-01-26 09:22:30
中国造纸(2020年9期)2020-10-20 05:33:36
公民与法治(2020年15期)2020-09-25 02:57:54
家庭医学(下半月)(2020年7期)2020-08-24 07:47:04
四川环境(2019年6期)2019-03-04 09:48:50
知识经济·中国直销(2018年1期)2018-01-31 01:52:37
商周刊(2017年6期)2017-08-22 03:42:37
高原山地气象研究(2016年2期)2016-11-10 06:06:27
